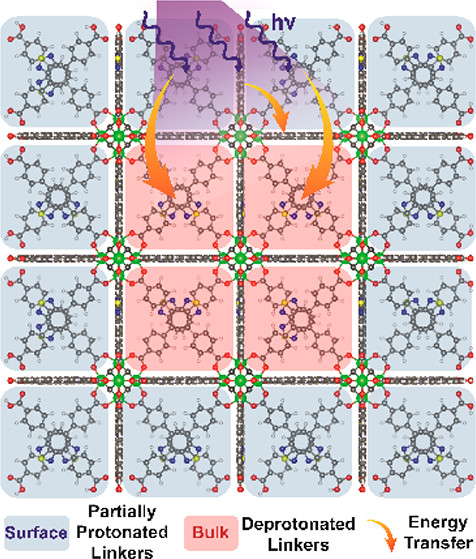
Highly luminescent metal–organic frameworks (MOFs) have recently received great attention due to their potential applications as sensors and light-emitting devices. In these MOFs, the highly ordered fluorescent organic linkers positioning prevents excited-state self-quenching and rotational motion, enhancing their light-harvesting properties. Here, the exciton migration between the organic linkers with the same chemical structure but different protonation degrees in Zr-based MOFs was explored and deciphered using ultrafast laser spectroscopy and density functional theory calculations. First, we clearly demonstrate how hydrogen-bonding interactions between free linkers and solvents affect the twisting changes, internal conversion processes, and luminescent behavior of a benzoimidazole-based linker. Second, we provide clear evidence of an ultrafast energy transfer between well-aligned adjacent linkers with different protonation states inside the MOF. These findings provide a new fundamental photophysical insight into the exciton migration dynamics between linkers with different protonation states coexisting at different locations in MOFs and serve as a benchmark for improving light-harvesting MOF architectures.