
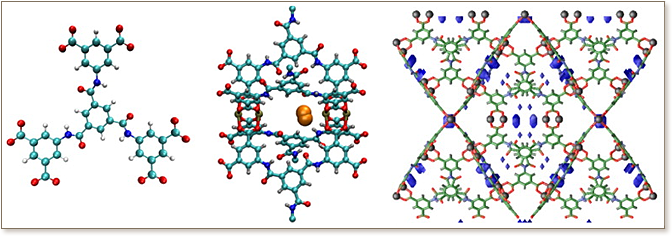
 Grand canonical Monte Carlo (GCMC) studies of the mechanism of hydrogen sorption in an rht-MOF known as Cu-TPBTM are presented. The MOF is a decorated/substituted isostructural analogue to the unembellished rht-MOF, PCN-61, that was studied previously [Forrest, K. A. et al. J. Phys. Chem. C 2012, 116, 15538-15549.]. The simulations were performed using three different hydrogen potentials of increasing complexity. Simulated hydrogen sorption isotherms and calculated isosteric heat of adsorption, Q values were in excellent agreement with the reported experimental data for only a polarizable model in one of four experimentally observed crystal structure configurations. The study demonstrates the ability of modeling to distinguish the differential sorption of distinct strucures; one configuration is found to be dominant due to favorable interactions with substrates. In addition, it was discovered that the presence of polar amide groups had a significant effect on the electrostatics of the Cu2+ ions and directs the low-pressure physisorption of hydrogen in the MOF. This is in contrast to what was observed in PCN-61, where an exterior copper ion had a higher relative charge and was the favored loading site. This tunability of the electrostatics of the copper ions via chemical substitution on the MOF framework can be explained by the presence of the negatively charged oxygen atom of the amide group that causes the interior Cu2+ ion to exhibit a higher positive charge through an inductive effect. Further, control simulations, taking advantage of the flexibility afforded by theoretical modeling, include artificially modified charges for both Cu2+ ions chosen equal to or with a higher charge on the exterior Cu2+ ion. This choice resulted in distinctly different hydrogen sorption characteristics in Cu-TPBTM with no direct sorption on the open-metal sites. Thus, this study demonstrates both the tunable nature of MOF platforms and the possibility for rational design of sorption/catalytic sites and characteristics through the active interplay of theory and experiment. The ability of accurate, carefully parametrized and transferable force fields to model and predict small molecule sorption in MOFs, even including open-metal sites, is demonstrated.
Grand canonical Monte Carlo (GCMC) studies of the mechanism of hydrogen sorption in an rht-MOF known as Cu-TPBTM are presented. The MOF is a decorated/substituted isostructural analogue to the unembellished rht-MOF, PCN-61, that was studied previously [Forrest, K. A. et al. J. Phys. Chem. C 2012, 116, 15538-15549.]. The simulations were performed using three different hydrogen potentials of increasing complexity. Simulated hydrogen sorption isotherms and calculated isosteric heat of adsorption, Q values were in excellent agreement with the reported experimental data for only a polarizable model in one of four experimentally observed crystal structure configurations. The study demonstrates the ability of modeling to distinguish the differential sorption of distinct strucures; one configuration is found to be dominant due to favorable interactions with substrates. In addition, it was discovered that the presence of polar amide groups had a significant effect on the electrostatics of the Cu2+ ions and directs the low-pressure physisorption of hydrogen in the MOF. This is in contrast to what was observed in PCN-61, where an exterior copper ion had a higher relative charge and was the favored loading site. This tunability of the electrostatics of the copper ions via chemical substitution on the MOF framework can be explained by the presence of the negatively charged oxygen atom of the amide group that causes the interior Cu2+ ion to exhibit a higher positive charge through an inductive effect. Further, control simulations, taking advantage of the flexibility afforded by theoretical modeling, include artificially modified charges for both Cu2+ ions chosen equal to or with a higher charge on the exterior Cu2+ ion. This choice resulted in distinctly different hydrogen sorption characteristics in Cu-TPBTM with no direct sorption on the open-metal sites. Thus, this study demonstrates both the tunable nature of MOF platforms and the possibility for rational design of sorption/catalytic sites and characteristics through the active interplay of theory and experiment. The ability of accurate, carefully parametrized and transferable force fields to model and predict small molecule sorption in MOFs, even including open-metal sites, is demonstrated.