
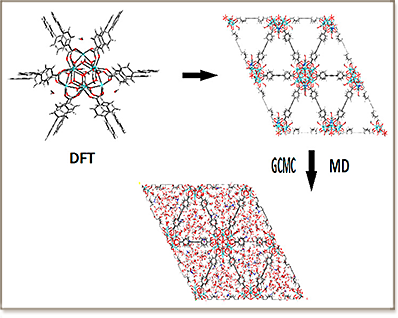
 The adsorption of water in the recently synthesized rare-earthbased Y-shp-MOF-5 metal−organic framework was explored by combining density functional theory calculations and force-field-based Monte Carlo and molecular dynamics simulations in order to unravel the atomistic mechanism that controls the high water uptake in this hydrolytically stable porous material. These simulations revealed that the strong hydrogen-bonding interaction of water with the extraframework dimethylammonium cations (DMA+ ) is one of the main driving forces for the high adsorption uptake of water. The terminal −(OH) groups bound to the rare earth sites as well as the aromatic ring of the organic linkers make the MOF−H2O interactions stronger leading to a high water affinity of Y-shp-MOF-5, and hence a large water uptake in the pores. The dynamics related to the local structural order and the hydrogen-bonding network around the adsorbed water molecules were also studied, revealing a significant slowing-down process compared to the corresponding dynamics of the bulk liquid phase. It was further demonstrated that the presence of strong hydrogen bonds drastically affects the translational and rotational dynamics of the confined water molecules and DMA+ cations, as reflected on the behavior of the corresponding time correlation functions at short and intermediate time scales.
The adsorption of water in the recently synthesized rare-earthbased Y-shp-MOF-5 metal−organic framework was explored by combining density functional theory calculations and force-field-based Monte Carlo and molecular dynamics simulations in order to unravel the atomistic mechanism that controls the high water uptake in this hydrolytically stable porous material. These simulations revealed that the strong hydrogen-bonding interaction of water with the extraframework dimethylammonium cations (DMA+ ) is one of the main driving forces for the high adsorption uptake of water. The terminal −(OH) groups bound to the rare earth sites as well as the aromatic ring of the organic linkers make the MOF−H2O interactions stronger leading to a high water affinity of Y-shp-MOF-5, and hence a large water uptake in the pores. The dynamics related to the local structural order and the hydrogen-bonding network around the adsorbed water molecules were also studied, revealing a significant slowing-down process compared to the corresponding dynamics of the bulk liquid phase. It was further demonstrated that the presence of strong hydrogen bonds drastically affects the translational and rotational dynamics of the confined water molecules and DMA+ cations, as reflected on the behavior of the corresponding time correlation functions at short and intermediate time scales.